Аномалия Эбштейна
- Аннотация
- Статья
- Ссылки
- English
В качестве возможных причин развития порока называют генетические и средовые факторы (вирусные инфекции, применение бензодиазепинов, препаратов лития). Клинически заболевание проявляется цианозом, одышкой, сердечной недостаточностью, нарушением ритма сердца. Морфологически отмечаются аномалии трикуспидального клапана, сосочковых мышц и хордального аппарата, гипертрофия миокарда правого желудочка, истончение стенок правого предсердия и атриализованной части правого желудочка. Решающим для диагноза является ультразвуковое исследование, по данным которого обнаруживают смещение правого атриовентрикулярного кольца. Степень смещения определяет тяжесть порока. Амплитуда движения трикуспидального клапана увеличена. Закрытие этого клапана в отличие от митрального происходит позднее. Разница 40 мс между точками закрытия трикуспидального и митрального клапанов – диагностически важный признак аномалии Эбштейна. Тактика врача определяется клинической картиной.
В качестве возможных причин развития порока называют генетические и средовые факторы (вирусные инфекции, применение бензодиазепинов, препаратов лития). Клинически заболевание проявляется цианозом, одышкой, сердечной недостаточностью, нарушением ритма сердца. Морфологически отмечаются аномалии трикуспидального клапана, сосочковых мышц и хордального аппарата, гипертрофия миокарда правого желудочка, истончение стенок правого предсердия и атриализованной части правого желудочка. Решающим для диагноза является ультразвуковое исследование, по данным которого обнаруживают смещение правого атриовентрикулярного кольца. Степень смещения определяет тяжесть порока. Амплитуда движения трикуспидального клапана увеличена. Закрытие этого клапана в отличие от митрального происходит позднее. Разница 40 мс между точками закрытия трикуспидального и митрального клапанов – диагностически важный признак аномалии Эбштейна. Тактика врача определяется клинической картиной.
Определение
Аномалия Эбштейна (Q 22.5) – редкий врожденный порок, заключающийся в апикальном смещении правого атриовентрикулярного кольца с аномалией развития трикуспидального клапана и нередко сосуществованием других пороков сердца [1]. Порок впервые был описан Вильгельмом Эбштейном в 1866 г. у 19-летнего юноши с одышкой, тахикардией, цианозом, расширением яремных вен и границ сердца.
Аномалия Эбштейна часто сочетается с дефектом межпредсердной перегородки (90% всех случаев), стенозом или атрезией легочной артерии (20–25%), синдромом Вольффа – Паркинсона – Уайта (20%). При лево-транспозиции магистральных сосудов (L-TGA) трикуспидальный клапан расположен слева, что говорит о эбштеноидной аномалии [2].
Распространенность и возможные причинные факторы
Аномалия Эбштейна регистрируется с частотой 1:200 000 живорожденных, составляя 0,3–0,6% всех случаев врожденных пороков сердца. Мы наблюдали шесть пациентов с аномалией Эбштейна. По нашим данным, за 42 года работы патологоанатомического отделения областной больницы этот порок диагностирован в двух случаях (более чем на 6000 секционных исследований) – у взрослого и ребенка. За период 1979–2019 гг. в кабинетах ультразвуковой диагностики многопрофильных детских больниц аномалия Эбштейна выявлена нами в двух случаях на более чем 32 000 эхокардиографий. В городской детской поликлинике зафиксировано еще два случая на 24 000 эхокардиографий. У наших пациентов аномалия Эбштейна сочеталась с дефектом межпредсердной перегородки (4/6) и синдромом Вольффа – Паркинсона – Уайта (1/6). Во всех наших наблюдениях имели место спорадические варианты. В литературе описаны семейные варианты, хотя подчеркивается спорадичность подавляющего большинства случаев. Высокая частота аномалии Эбштейна (6:100) зарегистрирована у детей, рожденных матерями с тем же пороком. Если носителем аномалии является отец, вероятность рождения ребенка с аномалией Эбштейна снижается в десять раз (6:1000). Тем не менее она значительно выше, чем в общей популяции.
Специфического гена, ответственного за развитие аномалии Эбштейна, не выявлено. Генами-кандидатами являются MYH7 и NKX2-5. У носителей порока нередко выявляется миссенс-мутация FLNA (филамин А, актин-связывающий протеин) на Xq28 [3–6]. У родственников редко обнаруживаются мутации кардиального фактора транскрипции NKX2-5, делеции 10p13-p14 и 1p34.3-p36.11 [7]. В качестве возможных внешних факторов называют вирусные инфекции, применение беременными бензодиазепинов и препаратов лития [8]. Последний считают ответственным менее чем за 2% случаев аномалии Эбштейна [9].
Патоморфология и нарушения гемодинамики
В здоровом сердце правое и левое атриовентрикулярные кольца расположены на одном уровне. Трехстворчатый клапан состоит из трех лепестков (передняя, задняя и септальная створки). Аномалия Эбштейна – нарушение развития трехстворчатого клапана и правого желудочка [10]:
- в период эмбрионального развития нарушается отслаивание зачатка створки клапана от миокарда. Бóльшей своей частью она остается спаянной с миокардом;
- правое атриовентрикулярное кольцо смещается в сторону верхушки сердца (септальная створка > задней > передней);
- часть правого желудочка, оказавшаяся над клапанным кольцом, расширяется, принимает на себя функцию предсердия (атриализуется), стенки атриализованной части истончаются;
- расширяется правое атриовентрикулярное кольцо.
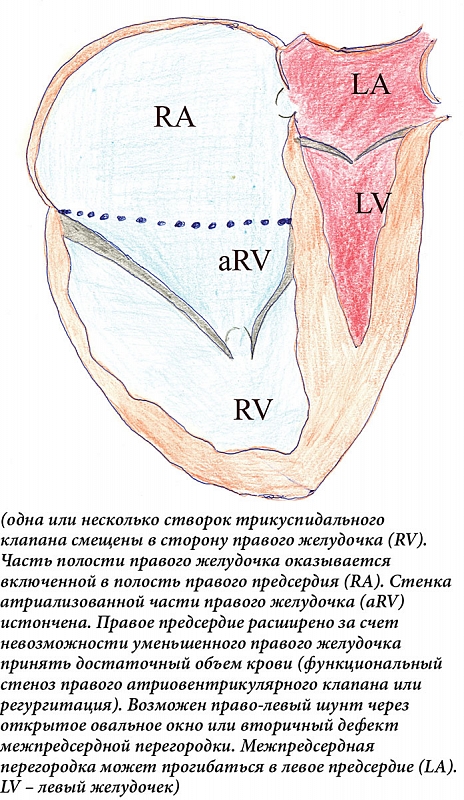
Точка максимального смещения находится на комиссуре между задней и септальной створками. Передняя створка увеличена в размерах, может быть перфорирована, деформирована. Несбалансированная деформация приводит к вращательному смещению трикуспидального клапана в выносной тракт правого желудочка, вызывая обструкцию. Хордальные нити передней створки укорочены, развиты плохо. Правый желудочек разделен на две части – вовлеченную в порок (атриализованную) часть правого желудочка и собственно правый желудочек в виде трабекулярной зоны и выносного канала. Атриализованная часть желудочка (входной канал) расширена, может составлять от трети (чаще) до половины (в тяжелых случаях) объема желудочка. Расширяется не только атриализованная часть, но и функциональная часть правого желудочка. Она может сдавливать левый желудочек вплоть до эпизодов обструкции выносного тракта левого желудочка [11, 12] (рис. 1).
По результатам анатомо-гистологического исследования, у трехмесячного ребенка выявлено резкое увеличение размера и массы (50 г) сердца. Правое фиброзное атриовентрикулярное кольцо смещено вниз и в сагиттальной плоскости, его правая сторона расположена у верхушки, а задняя и передняя стороны – вертикально. Правое предсердие выглядит как большая тонкостенная полость (5,5 × 4,5 см) с толщиной стенки 0,1 см. На задней стенке правого желудочка находится неровный эндокардиальный валик, расположенный вертикально между истинной предсердно-желудочковой границей и верхушкой сердца. В таком же направлении к передней стенке правого желудочка прикрепляется единственный передний парус. Эндокардиальный валик и передний парус делят правый желудочек на две части – правую, непосредственное продолжение правого предсердия, с толщиной стенки 0,1 см и узкую левую – выносной тракт желудочка. Внутренняя поверхность правого предсердия и атриализованной части правого желудочка гладкая, лишена трабекулярных мышц. В области верхушки правого желудочка расположены две короткие тонкие сосочковые мышцы. От их верхушек отходят тонкие хордальные нити, которые крепятся к левой стороне переднего паруса и образовывают на нем беспорядочные сплетения. Овальное окно открыто. Микроскопически – в миокарде правого желудочка выраженный отек межмышечных пространств, единичные инфильтраты из гистиоцитов. Субэндокардиально обнаруживаются микронекрозы.
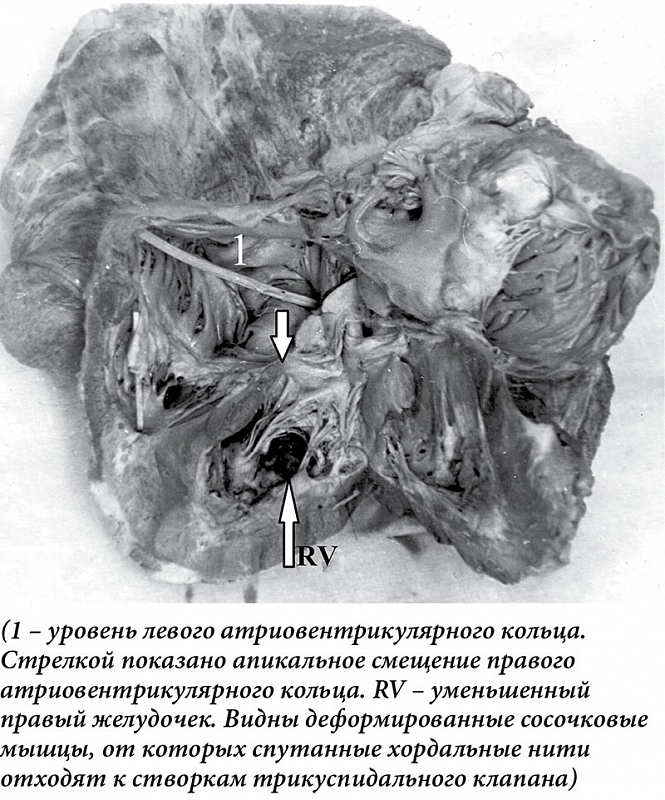
У мужчины 62 лет, занятого физическим трудом (каменщик), доставленного в стационар с манифестными признаками сердечной недостаточности, при аутопсии обнаружена жидкость в серозных полостях. Масса (470 г) и размеры сердца увеличены за счет правых отделов. Правое фиброзное кольцо смещено к верхушке желудочка. К кольцу прикрепляются три паруса трикуспидального клапана. Правое предсердие увеличено, тонкостенное. Правый желудочек уменьшен, от фиброзного кольца до верхушки – 3,2 см, ширина желудочка под фиброзным кольцом – 3,6 см. Стенки правого желудочка утолщены до 1,5 на уровне фиброзного кольца и 3,5 см – в области верхушки. В верхушке желудочка имеются две короткие толстые сосочковые мышцы (передняя и задняя). От передней мышцы хордальные нити тянутся к переднему парусу трикуспидального клапана, от задней – к заднему и медиальному. Хордальные нити тонкие, короткие. Эндокард утолщен, белесоватый. Фиброзные тяжи от эндокарда проникают на всю толщину миокарда правого желудочка. Выражена гипертрофия межжелудочковой перегородки и стенки левого желудочка. Гистологически – эндокард правого желудочка резко утолщен за счет разрастания коллагеновых волокон. Мышечные волокна правого желудочка гипертрофированы. Между ними широкие прослойки зрелой соединительной ткани. Коллагеновые волокна расположены параллельно, среди них немногочисленные клетки типа фибробластов. Широкие тяжи от эндокарда глубоко проникают в миокард, образуя поля склероза. В очагах склероза – атрофированные мышечные пучки (рис. 2).
Гемодинамика определяется выраженностью аномалии. Минимальное смещение правого атриовентрикулярного кольца при умеренной регургитации может длительное время оставаться без клинически значимых последствий. Существенное смещение клапанного кольца с выраженной недостаточностью трикуспидального клапана, значительным расширением полости правого предсердия и соответственно уменьшением полости правого желудочка приводит к декомпенсации, иногда уже у плода. Размеры функционально активного правого желудочка (за исключением его атриализованной части) могут быть настолько малы, что не способны обеспечить достаточное систолическое давление. Кроме того, имеет место недостаточность трехстворчатого клапана. Цианоз обусловлен тремя причинами:
- дефектом межпредсердной перегородки или открытым овальным окном;
- снижением кровотока через легкие при функциональном или анатомическом стенозе клапана легочной артерии;
- высоким сопротивлением сосудов легкого у новорожденных.
Состояние несколько смягчает открытый артериальный проток, по крайней мере до снижения сопротивления легочных сосудов при перестройке плодного типа кровообращения. В дальнейшем возможно развитие сердечной недостаточности не только из-за малого размера правого желудочка с крайне низким комплаенсом и регургитацией через правое атриовентрикулярное отверстие, но также из-за дисфункции и дисхронии левого желудочка, часто возникающей суправентрикулярной тахикардии – 25–50% всех пациентов. У многих из них обнаруживается синдром Вольффа – Паркинсона – Уайта [13, 14].
Клиническая картина
Клиническая картина полностью определяется степенью смещения правого атриовентрикулярного кольца и наличием сопутствующих пороков. Чем меньше смещение, тем позже манифестирует кардиальная симптоматика. Носители порока могут дожить до зрелого возраста без хирургического вмешательства [15], что зафиксировано и в нашем наблюдении. У всех пациентов, находившихся под нашим наблюдением, отмечались тахикардия, расширение границ сердца, одышка, застойные явления в легких и увеличение размеров печени. Аускультативно определялся грубый систолический шум в основании сердца и сосудов. Рентгенологически фиксировали кардиомегалию, легочную олигемию, расширение правого предсердия и правого силуэта сердца.
Клиническая картина аномалии Эбштейна зависит не только от степени смещения клапана, но и от возраста манифестации [16, 17]:
- плод: типичную эхографическую картину удается зарегистрировать у 86% пораженных, аритмию – у 5%;
- новорожденный (0–1 месяц): цианоз регистрируется у 74%, сердечная недостаточность – у 10% (отказ от еды, задержка развития), шум в сердце – у 9%;
- 2 месяца – 2 года: цианоз – 35% случаев, сердечная недостаточность – 43%, шум в сердце – 13%;
- 3 года – 10 лет: цианоз – 14%, сердечная недостаточность – 8%, аритмия – 12%, шум в сердце – 66%;
- 11–18 лет: цианоз – 13%, сердечная недостаточность – 13%, аритмия – 40%, шум в сердце – 33%;
- взрослые (старше 18 лет): цианоз – 4%, сердечная недостаточность – 26%, аритмия – 43%, шум в сердце – 13%, боль в области сердца – 20%, обморок – 6%.
При аномалии Эбштейна электрокардиограмма (ЭКГ) показывает острую высокую волну Р (признак расширения правого предсердия), отклонение электрической оси сердца вправо, блокаду правой ножки пучка Гиса. При наличии синдрома Вольффа – Паркинсона – Уайта – короткий интервал PR и волну дельта. Не исключена предсердная аритмия.
Эхокардиография – решающая методика диагностики. Цель ультразвукового исследования – описать анатомические характеристики порока, сопутствующих аномалий и гемодинамические нарушения.
В М-режиме трикуспидальный клапан лоцируется левее обычного. Передняя створка кажется аномально большой. Амплитуда движения клапана увеличена. Закрытие этого клапана в отличие от митрального происходит позднее, что обусловлено атриализацией правого желудочка и снижением его насосной функции. Разница 40 мс между точками закрытия трикуспидального и митрального клапанов – диагностически важный признак аномалии Эбштейна, хотя и не всегда регистрируется. При возможности одномоментной регистрации клапанов легочной артерии и аорты видно, что клапан легочной артерии открывается и закрывается позже, чем аортальный, что также отражает сниженную насосную функцию правого желудочка. Наиболее информативна 2D-эхокардиография, считающаяся золотым стандартом диагностики этого порока. Гемодинамические характеристики можно получить по результатам спектральной и цветной допплерографии. Оптимальны трансторакальное четырехкамерное сканирование, сканирование по парастернальным и субкостальным длинной и короткой осям. При обследовании плода патология трикуспидального клапана лучше всего выявляется при четырехкамерном сканировании. Чреспищеводная эхография оправданна при оперативном вмешательстве.
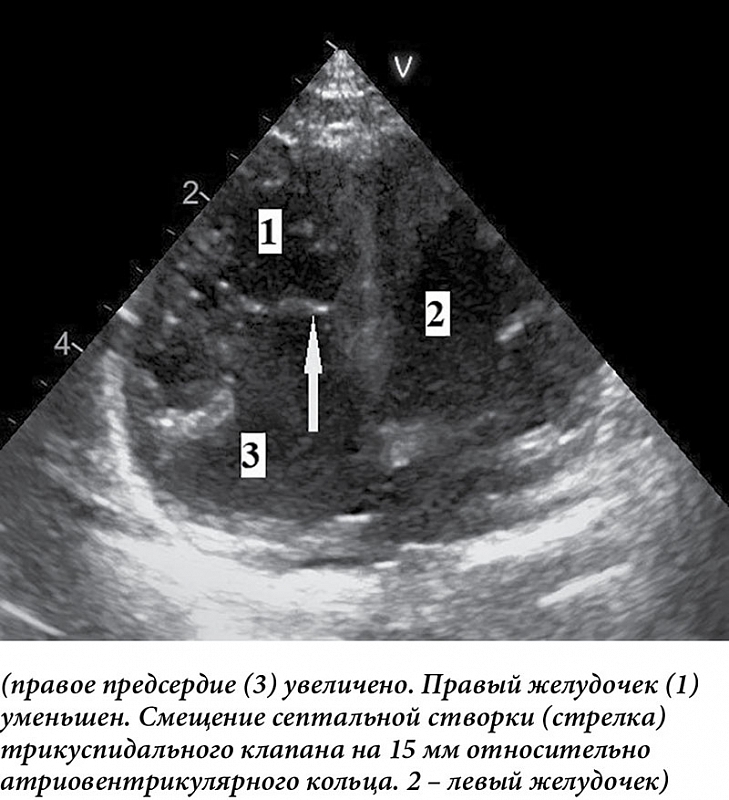
Апикальное смещение септальной створки (при тяжелых вариантах аномалии) относительно митрального клапана хорошо заметно на четырехкамерном изображении (рис. 3).
Увеличение правого предсердия оптимально оценивать при четырехкамерном сканировании, по парастернальной короткой оси и субкостально. Трикуспидальный клапан лучше всего лоцируется по парастернальной длинной и короткой осям, при четырехкамерном и субкостальном сканировании. По итогам такого сканирования можно определить степень тяжести аномалии, что важно для определения сроков оперативного вмешательства. Для этого при четырехкамерном сканировании в конце диастолы определяют частное от деления суммы площади правого предсердия (RA) и атриализованной части правого желудочка (aRV) на сумму площадей функциональной части правого желудочка (RV), левого предсердия (LA) и левого желудочка (LV): (RA + aRV)/(RV + LA + LV). Если полученная величина менее 0,5, говорят о первой степени тяжести (летальность 0%). Значение указанной величины 0,5–0,99 свидетельствует о второй степени (летальность до 10%), 1–1,49 – о третьей степени (летальность 44%), более 1,5 – четвертой степени (летальность практически 100%). Третья и четвертая степени тяжести – показатели высокой вероятности летального исхода [18–20]. В дальнейших исследованиях было показано, что не меньшее значение в качестве предикторов неблагоприятных исходов имеют дистресс плода, атрезия/стеноз легочной артерии [21].
Педиатрические проблемы ведения пациентов с аномалией Эбштейна
Из специалистов с ребенком чаще всего контактирует педиатр. Поэтому именно от него зависит, в какие сроки, к какому специалисту, на консультацию или диспансерное наблюдение будет направлен ребенок. Педиатр решает проблемы междисциплинарного и межпрофессионального взаимодействия.
Бессимптомные пациенты с аномалией Эбштейна с минимальной регургитацией в отсутствие других пороков развития и нарушения ритма сердца могут наблюдаться амбулаторно. В отсутствие клинических показаний (снижение аппетита и физической активности, плоская весовая кривая или переход показателей массы тела и роста на нижнюю перцентиль, одышка) один раз в 3–6 месяцев выполняется ЭКГ, один раз в шесть месяцев – эхокардиография и допплерография, определение сатурации кислорода. Консультация кардиолога проводится один раз в шесть месяцев, рентгенография – по показаниям.
В случае присоединения вирусной и/или бактериальной инфекции необходимо оценить показания к проведению ЭКГ и ультразвукового обследования сердца.
Профилактика бактериального эндокардита осуществляется пожизненно, вакцинация – по общему графику. Обязательна вакцинация от пневмо- и менингококка.
Диета – без особенностей. При тяжелых вариантах аномалии и признаках сердечной недостаточности назначают высококалорийные смеси. Физическая активность при аномалии Эбштейна зависит от степени смещения створок клапана, сопутствующих пороков или нарушений ритма сердца. Если смещение створок незначительное, нет пароксизмальной суправентрикулярной тахикардии, степень физической нагрузки определяет сам пациент [2]. Мы считаем целесообразным рекомендовать подготовительную группу по физкультуре. При наличии признаков недостаточности кровообращения, цианоза следует исключить занятия физкультурой в школе. Более точным критерием исключения или разрешения физических нагрузок и их степени являются стресс-тесты. Выполнять их можно только в специализированных условиях.
Появление признаков декомпенсации, цианоза требует немедленной консультации кардиолога и кардиохирурга. Хирургическое лечение пациентов с аномалией Эбштейна зависит от степени смещения створок, нарушения функций правого желудочка [22]. Новорожденные с тяжелой аномалией Эбштейна перед оперативным вмешательством для стабилизации состояния должны находиться в отделении интенсивной терапии. Новорожденным при недостаточности легочного кровотока или с дуктус-зависимым кровотоком вводится простагландин Е1.
Заключение
Аномалия Эбштейна характеризуется апикальным смещением правого атриовентрикулярного кольца из-за нарушенного отслоения задней и септальной створок от миокарда. Часто возникают недостаточность трикуспидального клапана, расширение правого предсердия и право-левый сброс через дефект межпредсердной перегородки. Средний возраст диагностики аномалии Эбштейна – 24 года. Один из наиболее частых клинических симптомов-дебютантов (50%) – аритмии [23].
Аномалия Эбштейна – редкое заболевание, чаще спорадическое. В казуистических случаях описывают семейные формы. Миссенс-мутация заключается (G>A) в замене электрически нейтрального (неполярного) глицина на положительно заряженный аргинин. Но фенотипический спектр этой мутации в FLNA чрезвычайно широк и связан прежде всего с аномалиями скелета. Указанная мутация отмечена при синдромах FG, фронтометафизальной дисплазии, синдроме Мельника – Нидлза, отопалатодигитальном синдроме типов I и II. Миссенс-мутация ассоциируется также с синдромами с неврологическими (перивентрикулярная нодулярная гетеротопия) и кардиальными (X-связанная дисплазия клапанов) нарушениями [24–27]. Но для аномалии Эбштейна указанные нарушения не характерны, то есть можно предположить, что мутация в FLNA – не единственная и не главная причина спорадической несиндромной аномалии Эбштейна.
Клиническая манифестация порока зависит от степени смещения трикуспидального клапана. Пациенты с признаками сердечной недостаточности, цианозом, расширением границ сердца должны быть незамедлительно направлены к кардиохирургу.
Решающим для диагноза аномалии Эбштейна является ультразвуковое исследование. В описании исследования должны быть отражены анатомия створок трикуспидального клапана, степень регургитации/стеноза, размеры правого предсердия и правого желудочка, сведения о шунтировании, функции миокарда правого и левого желудочков, а также всех сопутствующих нарушениях.
V.M. Delyagin, PhD, Prof., N.M. Doctorova, I.Ye. Belokrinitskaya, PhD
Dmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology
Scientific and Educational Biomedical Cluster ‘Translational Medicine’ Peoples' Friendship University of Russia
Contact person: Vasily M. Delyagin, delyagin-doktor@yandex.ru
Ebstein’s anomaly is a rare congenital malformation characterized by apical displacement of the right atrioventricular ring with an anomaly of the development of the tricuspid valve and the frequent presence of other heart defects. According to our data, this defect was diagnosed in 2 cases in more than 6 000 sectional studies: in an adult and a child. According to the echocardiography rooms of non-specialized multidisciplinary children's hospitals and children's polyclinics, the defect is detected with a frequency of 1:24.000–1:32.000 studies. Genetic and environmental factors (viral infections, benzodiazepines, lithium) are indicated as probable causes. Clinical signs are cyanosis, shortness of breath, heart failure, cardiac arrhythmias. Morphologically there are abnormalities of the tricuspid valve, papillary muscles and chordal apparatus, right ventricular myocardial hypertrophy, thinning of the walls of the right atrium and atrialized part of the right ventricle. Decisive for the diagnosis is ultrasound. Displacement of the right atrioventricular ring is revealed. The degree of bias determines the severity of the defect. The amplitude of the tricuspid valve movement is increased. Its valve closure occurs later than the mitral. A 40 ms difference between the tricuspid and mitral valve closure points is a diagnostically important sign of Ebstein’s anomaly. The tactics of a doctor are determined by the clinical picture.